- 599Nts data set

- 999Nts data set


Here we present the Robinson-Foulds metric computed for trees with 2133 and 200 Fungi taxa. Details on these datasets are presented at http://salsafungiphy.blogspot.com/2012/11/tree-distance-heatmaps.html
We have following five trees generated with this dataset.
| Tree A | Tree B | Tree C | Tree D | Tree E | |
| Tree A | 0 | 3484 | 3966 | 3726 | 3740 |
| Tree B | 3484 | 0 | 3956 | 3796 | 3806 |
| Tree C | 3966 | 3956 | 0 | 3438 | 3392 |
| Tree D | 3726 | 3796 | 3438 | 0 | 1282 |
| Tree E | 3740 | 3806 | 3392 | 1282 | 0 |
| Sequence Set | Tree Generation Method | Euclidean Mapping | Edge Sum | Stress 10D (3D) |
Correlation Regular Tree |
Heat Map | Plots | 2D View | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3D (3D) | 10D (10D) | 10D (3D) | With Original Pid | With 3D MDS Map | Edge Sum (Newick) vs Origin PID | MI-MDS vs Full MDS Mapping |
MSA Strict PIDNG vs SWG PID |
MSA Full PIDNG vs SWG PID |
Rectangular Cladogram | Cuboid Cladogram | Spherical Phylogram | |||||
| 126 + 74 (200) sequences Edge Sum Comparison Figure |
1. Clustal Omega FastTree | Internal Node Interpolation | 10.02 | 13.26 | 15.01 | 0.0264 | -- | -- | Spherical Cuboid |
Rectangle |  |
 |
||||
| Full 3D Map | -- | 11.29 | 0.0204 | |||||||||||||
| 2. Clustal Omega Raxml | Internal Node Interpolation | 11.33 | 14.62 | 16.15 | 0.0247 | 0.79 | 0.749 | Spherical Cuboid |
Rectangle |  |
 |
|||||
| Full 3D Map | -- | 13.05 | 0.02 | |||||||||||||
| 3. Clustal W2 FastTree | Internal Node Interpolation | 11.16 | 14.68 | 16.67 | 0.0277 | -- | -- | Spherical Cuboid |
Rectangle |  |
 |
|||||
| Full 3D Map | -- | 13.4 | 0.0205 | |||||||||||||
| 4. Clustal W2 Raxml | Internal Node Interpolation | 11.99 | 15.08 | 16.17 | 0.025 | 0.825 | 0.78 | Spherical Cuboid |
Rectangle |  |
 |
|||||
| Full 3D Map | -- | 13.69 | 0.0201 | |||||||||||||
| 5. Muscle FastTree | Internal Node Interpolation | 10.61 | 13.8 | 15.79 | 0.0274 | -- | -- | Spherical Cuboid |
Rectangle |  |
 |
|||||
| Full 3D Map | -- | 12.31 | 0.0202 | |||||||||||||
| 6. Muscle Raxml | Internal Node Interpolation | 10.90 | 14.67 | 16.44 | 0.027 | 0.823 | 0.775 | Spherical Cuboid |
Rectangle |  |
 |
|||||
| Full 3D Map | -- | 12.95 | 0.0202 | |||||||||||||
| 7. Neighbor Joining 3D | Internal Node Interpolation | 7.32 | 13.35 | -- | -- | -- | -- | n/a | n/a | Spherical | -- |  |
||||
| Full 3D Map | 7.33 | -- | -- | -- | ||||||||||||
| 8. Ninja 3D plot | Internal Node Interpolation | 7.36 | 12.92 | 14.51 | 0.0268 | 0.787 | 0.774 | n/a | n/a | Spherical Cuboid |
Rectangle |  |
 |
|||
| Full 3D Map | -- | 10.04 | 0.0198 | |||||||||||||
| 9. Neighbor Joining 10D | Internal Node Interpolation | 9.40 | 11.42 | 13.98 | 0.0285 | -- | -- | n/a | n/a | Spherical | -- |  |
||||
| Full 3D Map | -- | 9.9 | 0.0202 | |||||||||||||
| 10. Ninja 10D plot | Internal Node Interpolation | 8.65 | 12.13 | 14.99 | 0.0283 | 0.906 | 0.834 | n/a | n/a | Spherical Cuboid |
Rectangle |  |
 |
|||
| Full 3D Map | -- | 10.47 | 0.0207 | |||||||||||||
| 11. Ninja Original Pid | Internal Node Interpolation | 9.74 | 13.03 | 15.11 | 0.0273 | 0.925 | 0.855 |  |
n/a | n/a | Spherical Cuboid |
Rectangle |  |
 |
||
| -- | 11.67 | 0.0203 | ||||||||||||||
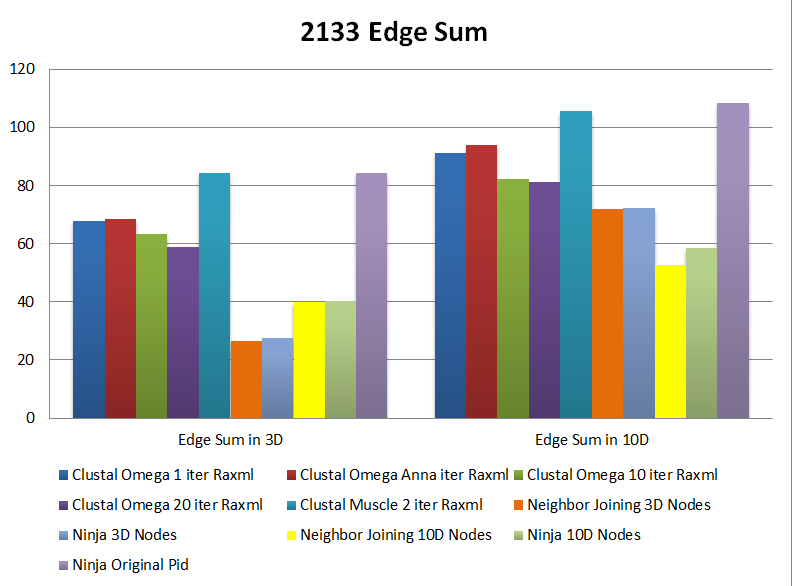
| 126 + 74 + 988 + 945 (2133) sequences Edge Sum Comparison Figure |
A. Clustal Omega 1 iter Raxml | Internal Node Interpolation | 67.95 | 91.31 | 99.9 | 0.025 | 0.138 | 0.145 | Spherical Cuboid |
 |
 |
|||||
| Full 3D Map | -- | 90.76 | 0.0229 | |||||||||||||
| B. Clustal Omega 1 iter FastTree | Internal Node Interpolation | 71.11 | 104.65 | -- | -- | Same as A | Same as A | Spherical Cuboid |
 |
 |
||||||
| Full 3D Map | -- | |||||||||||||||
| C. Clustal Omega Anna Raxml | Internal Node Interpolation | 68.34 | 93.91 | 102.23 | 0.025 | 0.679 | 0.714 | Spherical Cuboid |
 |
 |
||||||
| Full 3D Map | -- | 93.18 | 0.0226 | |||||||||||||
| D. Clustal Omega 10 iter Raxml | Internal Node Interpolation | 63.51 | 82.1 | 96.95 | 0.0262 | 0.272 | 0.284 | Spherical Cuboid |
Rectangle |  |
 |
|||||
| Full 3D Map | -- | 82.25 | 0.0232 | |||||||||||||
| E. Clustal Omega 20 iter Raxml | Internal Node Interpolation | 58.84 | 81.09 | 93.64 | 0.0252 | 0.275 | 0.288 | Spherical Cuboid |
Rectangle |  |
 |
|||||
| Full 3D Map | -- | 80.88 | 0.0229 | |||||||||||||
| F. Muscle 2 iter Raxml | Internal Node Interpolation | 84.23 | 105.52 | 113.82 | 0.0255 | 0.422 | 0.451 | Spherical Cuboid |
Rectangle |  |
 |
|||||
| Full 3D Map | -- | 103.03 | 0.0229 | |||||||||||||
| G. Neighbor Joining 3D | Internal Node Interpolation | 26.59 | 71.89 | -- | -- | -- | -- | n/a | n/a | Spherical | -- |  |
||||
| Full 3D Map | -- | -- | -- | |||||||||||||
| H. Ninja 3D plot | Internal Node Interpolation | 27.59 | 72.12 | 86.45 | 0.0268 | 0.519 | 0.575 | n/a | n/a | Spherical Cuboid |
Rectangle |  |
 |
|||
| Full 3D Map | -- | 66.61 | 0.023 | |||||||||||||
| I. Neighbor Joining 10D | Internal Node Interpolation | 39.85 | 52.56 | 81.68 | 0.028 | -- | -- | n/a | n/a | Spherical | -- |  |
||||
| Full 3D Map | -- | 61.16 | 0.0233 | |||||||||||||
| J. Ninja 10D transform | Internal Node Interpolation | 40.33 | 58.52 | 82.23 | 0.0264 | 0.728 | 0.781 | n/a | n/a | Spherical Cuboid |
Rectangle |  |
 |
|||
| Full 3D Map | -- | 63.41 | 0.0233 | |||||||||||||
| K. Ninja Original Pid | Internal Node Interpolation | 84.21 | 108.51 | 114.85 | 0.0247 | 0.614 | 0.605 |  |
n/a | n/a | Spherical Cuboid |
Rectangle |  |
 |
||
| Full 3D Map | -- | 106.83 | 0.0224 | |||||||||||||
| 3D mapping | 10D mapping | |
|---|---|---|
| 200 Sequences | 0.907 | 0.982 |
| 2133 Sequences | 0.804 | 0.885 |
{kind=link}
{kind=link}