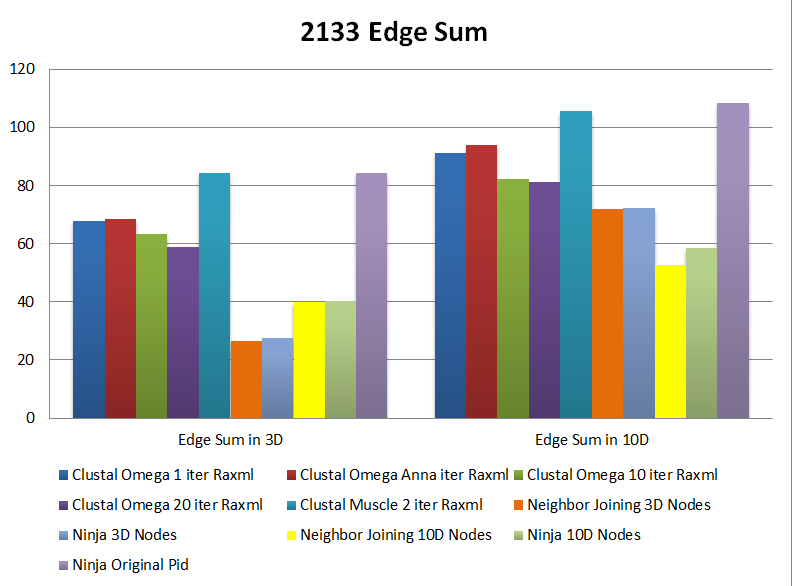
Here we present the Robinson-Foulds metric computed for trees with 2133 and 200 Fungi taxa. Details on these datasets are presented at http://salsafungiphy.blogspot.com/2012/11/tree-distance-heatmaps.html
Metric for Fungi 2133
We have following five trees generated with this dataset.
- Tree A
- Pairwise sequence alignment with Smith-Waterman algorithm using EDNAFULL scoring matrix and gap penalties –16 and –4 for gap open and gap extension respectively.
- Distance measure between sequences is percent identity
- Tree created with Ninja
- Tree B
- Multiple sequence alignment with MUSCLE
- Tree created with RAxML
- Tree C
- Distance between sequences are taken as the 3D distance resulted from DA-SMACOF dimensional scaling of the Smith-Waterman percent identity distances (used in Tree A).
- Tree created with Ninja
- Tree D
- Distance between sequences are taken as the 10D distance resulted from Manxcat (dimensional scaling program) running in SMACOF mode for the Smith-Waterman percent identity distances (used in Tree A).
- Tree created with Ninja
- Tree E
- Distance between sequences are taken as the 10D distance resulted from DA-SMACOF dimensional scaling of the Smith-Waterman percent identity distances (used in Tree A).
- Tree created with Ninja
Robinson-Foulds metric for trees
- The metric is computed with PHYLIP package found at http://evolution.genetics.washington.edu/phylip.html
| Tree A | Tree B | Tree C | Tree D | Tree E | |
| Tree A | 0 | 3484 | 3966 | 3726 | 3740 |
| Tree B | 3484 | 0 | 3956 | 3796 | 3806 |
| Tree C | 3966 | 3956 | 0 | 3438 | 3392 |
| Tree D | 3726 | 3796 | 3438 | 0 | 1282 |
| Tree E | 3740 | 3806 | 3392 | 1282 | 0 |










































{kind=link}
{kind=link}